
2310 ACIDITY*#(28)
2310 A. Introduction
Acidity of a water is its quantitative capacity to react with a strong base to a designated pH. The measured value may vary significantly with the end-point pH used in the determination. Acidity is a measure of an aggregate property of water and can be interpreted in terms of specific substances only when the chemcical composition of the sample is known. Strong mineral acids, weak acids such as carbonic and acetic, and hydrolyzing salts such as iron or aluminum sulfates may contribute to the measured acidity according to the method of determination.
Acids contribute to corrosiveness and influence chemical reaction rates, chemical speciation, and biological processes. The measurement also reflects a change in the quality of the source water.
2310 B. Titration Method
1. General Discussion
a. Principle: Hydrogen ions present in a sample as a result of dissociation or hydrolysis of solutes react with additions of standard alkali. Acidity thus depends on the end-point pH or indicator used. The construction of a titration curve by recording sample pH after successive small measured additions of titrant permits identification of inflection points and buffering capacity, if any, and allows the acidity to be determined with respect to any pH of interest.
In the titration of a single acidic species, as in the standardization of reagents, the most accurate end point is obtained from the inflection point of a titration curve. The inflection point is the pH at which curvature changes from convex to concave or vice versa.
Because accurate identification of inflection points may be difficult or impossible in buffered or complex mixtures, the titration in such cases is carried to an arbitrary end-point pH based on practical considerations. For routine control titrations or rapid preliminary estimates of acidity, the color change of an indicator may be used for the end point. Samples of industrial wastes, acid mine drainage, or other solutions that contain appreciable amounts of hydrolyzable metal ions such as iron, aluminum, or manganese are treated with hydrogen peroxide to ensure oxidation of any reduced forms of polyvalent cations, and boiled to hasten hydrolysis. Acidity results may be highly variable if this procedure is not followed exactly.
b. End points: Ideally the end point of the acidity titration should correspond to the stoichiometric equivalence point for neutralization of acids present. The pH at the equivalence point will depend on the sample, the choice among multiple inflection points, and the intended use of the data.
Dissolved carbon dioxide (CO2) usually is the major acidic component of unpolluted surface waters; handle samples from such sources carefully to minimize the loss of dissolved gases. In a sample containing only carbon dioxide-bicarbonates-carbonates, titration to pH 8.3 at 25°C corresponds to stoichiometric neutralization of carbonic acid to bicarbonate. Because the color change of phenolphthalein indicator is close to pH 8.3, this value generally is accepted as a standard end point for titration of total acidity, including CO2 and most weak acids. Metacresol purple also has an end point at pH 8.3 and gives a sharper color change.
For more complex mixtures or buffered solutions selection of an inflection point may be subjective. Consequently, use fixed end points of pH 3.7 and pH 8.3 for standard acidity determinations via a potentiometric titration in wastewaters and natural waters where the simple carbonate equilibria discussed above cannot be assumed. Bromphenol blue has a sharp color change at its end point of 3.7. The resulting titrations are identified, traditionally, as ‘‘methyl orange acidity’’ (pH 3.7) and ‘‘phenolphthalein’’ or total acidity (pH 8.3) regardless of the actual method of measurement.
c. Interferences: Dissolved gases contributing to acidity or alkalinity, such as CO2, hydrogen sulfide, or ammonia, may be lost or gained during sampling, storage, or titration. Minimize such effects by titrating to the end point promptly after opening sample container, avoiding vigorous shaking or mixing, protecting sample from the atmosphere during titration, and letting sample become no warmer than it was at collection.
In the potentiometric titration, oily matter, suspended solids, precipitates, or other waste matter may coat the glass electrode and cause a sluggish response. Difficulty from this source is likely to be revealed in an erratic titration curve. Do not remove interferences from sample because they may contribute to its acidity. Briefly pause between titrant additions to let electrode come to equilibrium or clean the electrodes occasionally.
In samples containing oxidizable or hydrolyzable ions such as ferrous or ferric iron, aluminum, and manganese, the reaction rates at room temperature may be slow enough to cause drifting end points.
Do not use indicator titrations with colored or turbid samples that may obscure the color change at the end point. Residual free available chlorine in the sample may bleach the indicator. Eliminate this source of interference by adding 1 drop of 0.1M sodium thiosulfate (Na2S2O3).
d. Selection of procedure: Determine sample acidity from the volume of standard alkali required to titrate a portion to a pH of 8.3 (phenolphthalein acidity) or pH 3.7 (methyl orange acidity of wastewaters and grossly polluted waters). Titrate at room temperature using a properly calibrated pH meter, electrically operated titrator, or color indicators.
Use the hot peroxide procedure (¶ 4a) to pretreat samples known or suspected to contain hydrolyzable metal ions or reduced forms of polyvalent cation, such as iron pickle liquors, acid mine drainage, and other industrial wastes.
Color indicators may be used for routine and control titrations in the absence of interfering color and turbidity and for preliminary titrations to select sample size and strength of titrant (¶4b).
e. Sample size: The range of acidities found in wastewaters is so large that a single sample size and normality of base used as titrant cannot be specified. Use a sufficiently large volume of titrant (20 mL or more from a 50-mL buret) to obtain relatively good volumetric precision while keeping sample volume sufficiently small to permit sharp end points. For samples having acidities less than about 1000 mg as calcium carbonate (CaCO3)/L, select a volume with less than 50 mg CaCO3 equivalent acidity and titrate with 0.02N sodium hydroxide (NaOH). For acidities greater than about 1000 mg as CaCO3/L, use a portion containing acidity equivalent to less than 250 mg CaCO3 and titrate with 0.1N NaOH. If necessary, make a preliminary titration
to determine optimum sample size and/or normality of titrant.
f. Sampling and storage: Collect samples in polyethylene or borosilicate glass bottles and store at a low temperature. Fill bottles completely and cap tightly. Because waste samples may be subject to microbial action and to loss or gain of CO2 or other gases when exposed to air, analyze samples without delay, preferably within 1 d. If biological activity is suspected analyze within 6 h. Avoid sample agitation and prolonged exposure to air.
2. Apparatus
a. Electrometric titrator: Use any commercial pH meter or electrically operated titrator that uses a glass electrode and can be read to 0.05 pH unit. Standardize and calibrate according to the manufacturer’s instructions. Pay special attention to temperature compensation and electrode care. If automatic temperature compensation is not provided, titrate at 25 ± 5°C.
b. Titration vessel: The size and form will depend on the electrodes and the sample size. Keep the free space above the sample as small as practicable, but allow room for titrant and full immersion of the indicating portions of electrodes. For conventional-sized electrodes, use a 200-mL, tall-form Berzelius beaker without a spout. Fit beaker with a stopper having three holes, to accommodate the two electrodes and the buret. With a miniature combination glass-reference electrode use a 125-mL or 250-mL erlenmeyer flask with a two-hole stopper.
.c. Magnetic stirrer
.d. Pipets, volumetric
e. Flasks, volumetric, 1000-, 200-, 100-mL
f. Burets, borosilicate glass, 50-, 25-, 10-mL
g. Polyolefin bottle, 1-L
3. Reagents
a. Carbon dioxide-free water: Prepare all stock and standard solutions and dilution water for the standardization procedure with distilled or deionized water that has been freshly boiled for 15 min and cooled to room temperature. The final pH of the water should be ≥ 6.0 and its conductivity should be <2 μmhos/cm.
b. Potassium hydrogen phthalate solution, approximately 0.05N: Crush 15 to 20 g primary standard KHC8H4O4 to about 100 mesh and dry at 120°C for 2 h. Cool in a desiccator. Weigh 10.0 ± 0.5 g (to the nearest mg), transfer to a 1-L volumetric flask, and dilute to 1000 mL.
c. Standard sodium hydroxide titrant, 0.1N: Prepare solution approximately 0.1N as indicated under Preparation of Desk Reagents (see inside front cover). Standardize by titrating 40.00 mL KHC8H4O4 solution (¶ 3b), using a 25-mL buret. Titrate to the inflection point (¶ 1a), which should be close to pH 8.7. Calculate normality of NaOH:
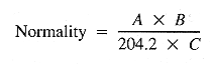
where:
A = g KHC8H4O4 weighed into 1-L flask,
B = mL KHC8H4O4 solution taken for titration, and
C = mL NaOH solution used.
Use the measured normality in further calculations or adjust to 0.1000N; 1 mL = 5.00 mg CaCO3.
d. Standard sodium hydroxide titrant, 0.02N: Dilute 200 mL 0.1N NaOH to 1000 mL and store in a polyolefin bottle protected from atmospheric CO2 by a soda lime tube or tight cap. Standardize against KHC8H4O4 as directed in ¶ 3c, using 15.00 mL KHC8H4O4 solution and a 50-mL buret. Calculate normality as above (¶ 3c); 1 mL = 1.00 mg CaCO3.
e. Hydrogen peroxide, H2O2, 30%.
f. Bromphenol blue indicator solution, pH 3.7 indicator: Dissolve 100 mg bromphenol blue, sodium salt, in 100 mL water.
g. Metacresol purple indicator solution, pH 8.3 indicator: Dissolve 100 mg metacresol purple in 100 mL water.
h. Phenolphthalein indicator solution, alcoholic, pH 8.3 indicator.
i. Sodium thiosulfate, 0.1M: Dissolve 25 g Na2S2O3⋅5H2O and dilute to 1000 mL with distilled water.
4. Procedure
If sample is free from hydrolyzable metal ions and reduced forms of polyvalent cations, proceed with analysis according to b, c, or d. If sample is known or suspected to contain such substances, pretreat according to a.
a. Hot peroxide treatment: Pipet a suitable sample (see ¶ 1e) into titration flasks. Measure pH. If pH is above 4.0 add 5-mL increments of 0.02N sulfuric acid (H2SO4) (Section 2320B.3c) to reduce pH to 4 or less. Remove electrodes. Add 5 drops 30% H2O2 and boil for 2 to 5 min.
Cool to room temperature and titrate with standard alkali to pH 8.3 according to the procedure of 4d.
b. Color change: Select sample size and normality of titrant according to criteria of ¶ 1e.
Adjust sample to room temperature, if necessary, and with a pipet discharge sample into an erlenmeyer flask, while keeping pipet tip near flask bottom. If free residual chlorine is present add 0.05 mL (1 drop) 0.1M Na2S2O3 solution, or destroy with ultraviolet radiation. Add 0.2 mL (5 drops) indicator solution and titrate over a white surface to a persistent color change characteristic of the equivalence point. Commercial indicator solutions or solids designated for the appropriate pH range (3.7 or 8.3) may be used. Check color at end point by adding the same concentration of indicator used with sample to a buffer solution at the designated pH.
c. Potentiometric titration curve:
1) Rinse electrodes and titration vessel with distilled water and drain. Select sample size and normality of titrant according to the criteria of ¶ 1e. Adjust sample to room temperature, if necessary, and with a pipet discharge sample while keeping pipet tip near the titration vessel bottom.
2) Measure sample pH. Add standard alkali in increments of 0.5 mL or less, such that a change of less than 0.2 pH units occurs per increment. After each addition, mix thoroughly but gently with a magnetic stirrer. Avoid splashing. Record pH when a constant reading is obtained.
Continue adding titrant and measure pH until pH 9 is reached. Construct the titration curve by plotting observed pH values versus cumulative milliliters titrant added. A smooth curve showing one or more inflections should be obtained. A ragged or erratic curve may indicate that equilibrium was not reached between successive alkali additions. Determine acidity relative to a particular pH from the curve.
d. Potentiometric titration to pH 3.7 or 8.3: Prepare sample and titration assembly as specified in ¶ 4c1). Titrate to preselected end-point pH (¶ 1d) without recording intermediate pH values. As the end point is approached make smaller additions of alkali and be sure that pH equilibrium is reached before making the next addition.
5. Calculation
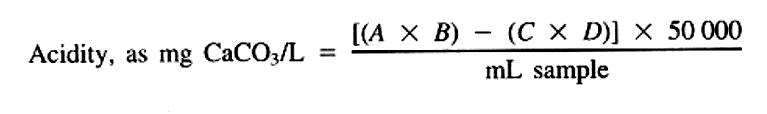
where:
A = mL NaOH titrant used,
B = normality of NaOH,
C = mL H2SO4 used (¶ 4a), and
D = normality of H2SO4.
Report pH of the end point used, as follows: ‘‘The acidity to pH _______ = _______ mg
CaCO3/L.’’ If a negative value is obtained, report the value as negative. The absolute value of this negative value should be equivalent to the net alkalinity.
6. Precision and Bias
No general statement can be made about precision because of the great variation in sample characteristics. The precision of the titration is likely to be much greater than the uncertainties involved in sampling and sample handling before analysis.
Forty analysts in 17 laboratories analyzed synthetic water samples containing increments of bicarbonate equivalent to 20 mg CaCO3/L. Titration according to the procedure of ¶ 4d gave a standard deviation of 1.8 mg CaCO3/L, with negligible bias. Five laboratories analyzed two samples containing sulfuric, acetic, and formic acids and aluminum chloride by the procedures of ¶s 4b and 4d.
The mean acidity of one sample (to pH 3.7) was 487 mg CaCO3/L, with a standard deviation of 11 mg/L. The bromphenol blue titration of the same sample was 90 mg/L greater, with a standard deviation of 110 mg/L. The other sample had a potentiometric titration of 547 mg/L, with a standard deviation of 54 mg/L, while the corresponding indicator result was 85 mg/L greater, with a standard deviation of 56 mg/L. The major difference between the samples was the substitution of ferric ammonium citrate, in the second sample, for part of the aluminum chloride.
7. Bibliography
WINTER, J.A. & M.R. MIDGETT. 1969. FWPCA Method Study 1. Mineral and Physical Analyses.
Federal Water Pollution Control Admin., Washington, D.C.
BROWN, E., M .W. SKOUGSTAD & M.J. FISHMAN. 1970. Methods for collection and analysis of water samples for dissolved minerals and gases. Chapter A1 in Book 5, Techniques of Water-Resources Investigations of United States Geological Survey. U.S. Geological Survey, Washington, D.C.
SNOEYINK, V.L. & D. JENKINS. 1980. Water Chemistry. John Wiley & Sons, New York, N.Y.


